
Scott Schrage, January 23, 2020
Enzymes and X-rays: Study reveals hidden acrobatics of cellular catalysts
After a two-year wait, they had two days.
Two days to fire historically intense X-rays at enzymes, the cellular turbo boosts that can shorten the duration of essential chemical reactions from 50 million years to 50 milliseconds.
Two days to observe, for the first time, an enzymatic gymnastic routine that had only been hypothesized.
Two days to show that their approach might help guide the design of pharmaceutical drugs that target cancers or Parkinson’s disease with greater precision and tenacity.
“Day one did not go well,” said Mark Wilson, associate professor of biochemistry at the University of Nebraska–Lincoln. “I remember going home and thinking, ‘Oh, God. This might be a wash. I really hope we do better tomorrow.’”
Thankfully for Wilson and his colleagues — biochemists, chemists and laser physicists stationed at universities ranging from California and Nebraska to Puerto Rico and Germany — tomorrow really was another day.
“I would have been happy if we saw what I expected,” Wilson said of day two. “I was delighted that we ended up seeing what we actually saw.”
What they saw — the temporary metamorphosis of an enzyme, the nanoscopic equivalent of a reversible mousetrap in action — could help resolve some longstanding and spirited debates in biochemistry.
‘That was a good day’
The team would eventually focus its scarce time and X-rays on isocyanide hydratase, an enzyme native to some bacterial species that need it to neutralize the toxic isocyanide molecules that can otherwise kill their cells. The enzyme is structurally similar to another that seems to drive diseases in humans, which is why Wilson initially took interest in it.
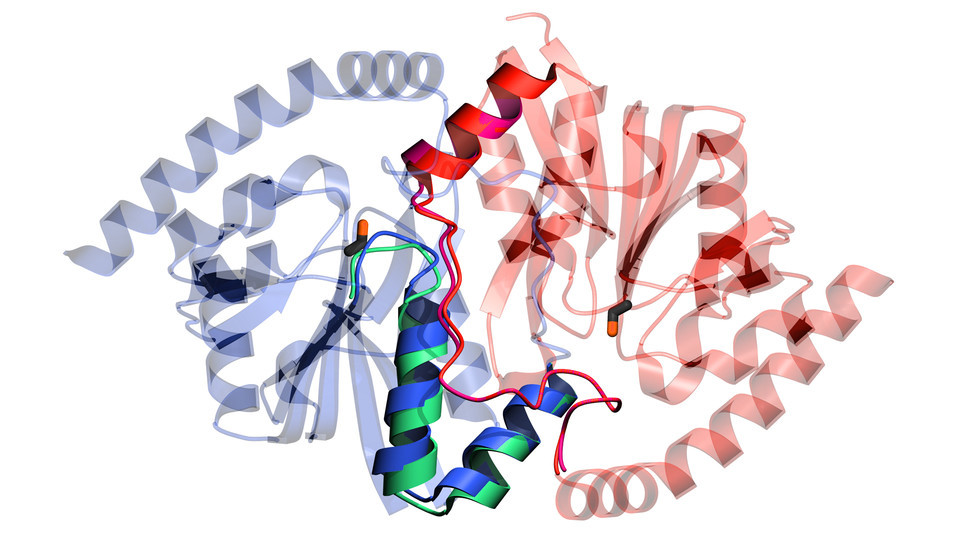
But the researchers came to realize that isocyanide hydratase might also help clarify an elusive question. Any biochemist worth their lab coat knows that enzymes are elastic and springy, allowing their amino acids — essentially the bones of their skeletal structures — to briefly shift position when encountering changes in temperature, pH or other conditions. What remains unclear: How much, if at all, do those reconfigurations actually help an enzyme accelerate chemical reactions in a cell?
“This has been very contentious,” Wilson said.
In pursuit of answers, Wilson and his colleagues turned to an emerging technology uniquely suited for the task: an X-ray free electron laser. The technology works by accelerating a beam of electrons to nearly the speed of light before sending it through a gauntlet of alternately polarized magnets, ultimately forcing the electrons to radiate X-rays that are roughly a billion times more intense than those produced via any previous approach. By recording how the X-rays diffract from their target, laser physicists can then calculate the structure of that target.
Wilson wanted to aim those X-rays at a slurry containing millions of microscopic crystals of isocyanide hydratase enzymes and the isocyanide molecules that they help detoxify. Doing so at various points after the two components had mixed — 10 milliseconds, 100 milliseconds, one second, for instance — would theoretically allow the team to capture high-resolution snapshots of the enzyme as it catalyzed a chemical reaction.
The problem? Only seven X-ray free electron lasers exist — the lone North American edition resides at Stanford University — and all are in high demand. That meant a two-year waiting list and, when that wait was over, just two days with the Linac Coherent Light Source at the SLAC National Accelerator Laboratory.
Going in, the researchers suspected that isocyanide hydratase might form a covalent bond with isocyanide — that the enzyme and its so-called substrate begin sharing a pair of electrons — as the former starts to act on the latter. They also speculated that this covalent bond formation could trigger a series of enzymatic motions essential to completing the chemical reaction.
By the end of the second day, the X-rays were confirming their suspicions: The covalent bond yielded another reactive but short-lived molecule, known as an intermediate, that was trapped within the enzyme. That intermediate then released the equivalent of a spring-loaded gate, weakening a hydrogen bond within isocyanide hydratase and granting water molecules access to its interior. Those water molecules latched onto and activated a particular site in the enzyme, kick-starting further chemical reactions needed to detoxify isocyanide. Eventually, the enzyme regained its tension, tightening back into its original configuration.
“It’s kind of like a mousetrap,” Wilson said. “When you set the mousetrap, the enzyme is strained. When the intermediate forms, all of a sudden, this part starts to move, which is important for getting the intermediate out. So the second half of the reaction requires the mobility that is allowed by the first half of the reaction.
“To the best of my knowledge, this is the first example of an enzyme whose otherwise hypothetical or unknown mechanism was established using this technique.”
Nearly as important as the findings themselves, Wilson said, were their detail and clarity. When Stanford’s Henry van den Bedem, the study’s co-leader, showed Wilson the first electron-density maps — similar to topographical maps that indicate the positions of atoms in a molecule — he was taken aback by the results.
“I expected it to be difficult to interpret,” Wilson said. “He showed me a picture that was unambiguously the intermediate — the thing we had postulated. You could see every atom. It was remarkably clear.
“That’s when I had the realization that this experiment had been far more successful than we had initially hoped. And those are kind of rare in science. I think they tend to get a lot of billing: ‘Oh, here’s the eureka moment!’ Usually, it’s a hard slog, and it’s very incremental. It’s a messy affair. But this was truly a moment when we realized this worked. So that was a good day.”
Per cysteine
One of isocyanide hydratase’s magic buttons — the amino acid that binds with isocyanide to kick-start the detoxifying chemical reaction and temporarily reconfigure the enzyme itself — is known as a cysteine. As the most highly reactive type of amino acid in biology, cysteines help promote chemical reactions in a wide range of enzymes and other cellular proteins.
Cysteine-related mutations in proteins native to the human body have been linked to some cancers, making them an attractive target for therapeutic drugs designed to halt their progression. And because the enzymes of foreign agents such as bacteria can facilitate infection, their cysteines are likewise appealing targets.
In revealing more about how cysteine-related reactions can reconfigure enzymes, the new study — and future research like it — should inform efforts to engineer drugs that can permanently inactivate troublesome enzymes by forming covalent bonds with some of those cysteines, Wilson said.
“Most drugs are aimed at enzymes,” he said, “and better understanding how enzymes function allows us to better target them with new generations of drugs (using) approaches that are directly relevant to the work we did.”
Wilson said he hopes that the study’s success will also encourage the global community of enzymologists to begin training X-ray free electron lasers on the enzymes, and the questions, that are most relevant to their own work.
“We’re still figuring out how best to do these experiments,” he said of his own ongoing work with collaborators throughout the United States. “We’re still trying to figure out what we can extract from them, but the initial results are very exciting. We think that this is a once-in-a-generation opportunity to use new technologies to answer longstanding questions in enzymology.”
The team reported its findings in the journal Proceedings of the National Academy of Sciences. Wilson and van den Bedem, who is now at Atomwise Inc., co-authored the study with Nebraska’s Medhanjali Dasgupta, David Berkowitz, Javier Seravalli, Virendra Tiwari, Peter Madzelan and doctoral alumnus Gregory Applegate, along with researchers from Stanford, Friedrich-Alexander University Erlangen-Nürnberg, the University of Puerto Rico, Lawrence Berkeley National Laboratory, the University of California, San Francisco, and Los Alamos National Laboratory.
The researchers received support in part from the U.S. Department of Energy, the National Science Foundation and the National Institutes of Health.




